Présentation de la maladie
Qu’est-ce que la protéinose alvéolaire pulmonaire ?
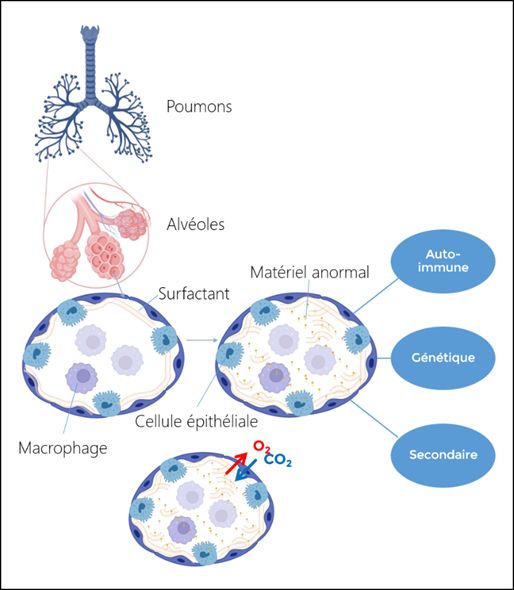
La protéinose alvéolaire pulmonaire (PAP) est une maladie respiratoire rare qui peut toucher les enfants et les adultes. Chez l’enfant, sa fréquence est estimée à quelques cas par million d’habitants. Cette maladie est liée à l’accumulation de matériel anormal dans les alvéoles pulmonaires qui sont normalement uniquement remplies d’air pour permettre les échanges gazeux à savoir la diffusion d’oxygène (O2) dans les vaisseaux sanguins pour oxygéner tous les organes de notre corps et l’épuration du dioxyde de carbone (CO2) à l’extérieur de notre organisme.
La principale conséquence de cette accumulation de matériel anormal est une altération des échanges gazeux car ce matériel va gêner la bonne diffusion des 2 gaz et entraîner une insuffisance respiratoire. Les symptômes de cette maladie découlent principalement de cette insuffisance respiratoire avec un essoufflement progressif, une fatigue et parfois une toux.
Les signes visibles peuvent être une respiration rapide avec des « signes de lutte » c’est-à-dire un creusement du thorax et des côtes lorsque l’enfant respire, une cyanose, c’est-à-dire une coloration bleutée de la peau autour de la bouche et au niveau des mains et des pieds lorsque la maladie entraîne un manque d’oxygène important et une déformation des extrémités des doigts et des orteils en baguette de tambour appelé « hippocratisme digital ».
Chez l’enfant, surtout le nourrisson, l’effort fourni pour la respiration peut entraîner des difficultés à bien manger et à bien grossir. Un retard de croissance est donc souvent un signe visible associé.
Quelles sont les origines et causes possibles de cette maladie ?
Comme indiqué sur la figure, les PAP peuvent être classées en 3 catégories en fonction de leur cause : auto-immune, génétique et secondaire. Ces formes ont toutes en commun une anomalie du recyclage du surfactant. Le surfactant est un film qui tapisse les alvéoles pulmonaires un peu comme une bulle de savon et qui permet que les alvéoles ne se ferment pas qu’on expire l’air. Ce film est fabriqué par les cellules épithéliales alvéolaires et est régulièrement recyclé pour être toujours propre et efficace. Ce recyclage est effectué par les macrophages alvéolaires qui vont l’avaler et le digérer, puis les cellules épithéliales en fabriquent une nouvelle couche, etc.
La forme auto-immune est une forme de l’adulte qui est exceptionnelle chez l’enfant. Les formes génétiques sont héréditaires et touchent des molécules qui sont importantes pour le bon fonctionnement du macrophage alvéolaire. En France, la forme génétique la plus répandue est celle liée à des mutations dans le gène MARS1 avec 2 nouveaux cas par an.
Enfin, les formes secondaires peuvent compliquer des maladies qui touche aussi le macrophage comme certaines leucémies, certaines maladies inflammatoires générales, des infections, des causes toxiques ou métaboliques.
Comment le diagnostic est-il établi ?
Comme indiqué dans la première partie, les signes respiratoires que peut présenter votre enfant ne sont pas spécifiques et ne font pas tout de suite penser à cette maladie. Dans certaines causes, notamment les mutations du gène MARS1, certains symptômes et signes extra-respiratoires associés peuvent évoquer la maladie comme une atteinte du foie, une diarrhée et des vomissements, et des épisodes de fièvre sans infection. Devant une insuffisance respiratoire chronique de l’enfant, le premier examen qui va nous orienter est le scanner pulmonaire qui va montrer des images typiques de la PAP qu’on appelle « crazy-paving ».
Cependant, cela ne suffit pas à confirmer le diagnostic et c’est l’analyse au microscope du liquide prélevé dans les poumons (lavage broncho-alvéolaire) lors d’une endoscopie bronchique (exploration des bronches avec un caméra) qui va permettre de poser le diagnostic avec certitude. L’aspect à l’œil nu du liquide prélevé est aussi souvent très caractéristique avec une couleur et une consistance laiteuses. Une fois le diagnostic confirmé, il faut en chercher la cause.
Quelles complications peuvent survenir ?
La principale complication est l’insuffisance respiratoire nécessitant une supplémentation en oxygène. Si cette insuffisance respiratoire n’est pas prise en charge, elle peut conduire au décès. La dénutrition et le retard de croissance sont des complications fréquentes. Enfin, les infections pulmonaires sont plus fréquentes chez les enfants atteints.
Quels traitements existent pour la protéinose alvéolaire pulmonaire ?
Les traitements de la PAP dépendent de la cause de la maladie chez chaque patient. En première intention, lorsque l’insuffisance respiratoire est importante, notamment quand l’enfant a besoin d’oxygène en continu ou a beaucoup de mal à manger et à grossir car il est très essoufflé, un lavage broncho-alvéolaire dit « thérapeutique » peut aider à libérer les alvéoles du matériel anormal et améliorer rapidement la respiration de l’enfant. Ce lavage consiste à laver chaque poumon en injectant du sérum physiologique lors d’une endoscopie sous anesthésie générale et de ré-aspirer le liquide injecté, un peu comme un lavage de nez lorsqu’un enfant est enrhumé mais en très grande quantité dans chaque poumon.
Dans certaines formes pour lesquelles une cause n’a pas été retrouvée et/ou lorsque la cause n’a pas de traitement spécifique, ces lavages sont le principal traitement proposé avec un rythme personnalisé adapté à chaque enfant. Ces lavages sont associés à une prise en charge plus générale de l’insuffisance respiratoire comme une supplémentation en oxygène soit continue soit la nuit et/ou pendant les efforts, et souvent une assistance nutritionnelle qui peut prendre la forme d’un enrichissement calorique des repas, des compléments hypercaloriques prêts à l’emploi ou une nutrition entérale par sonde nasogastrique ou gastrostomie dans les formes les plus sévères. Certaines causes disposent d’un traitement spécifique. C’est le cas de la protéinose MARS1 qui est traitée par de la méthionine par voie orale.
Une autre forme génétique, liée à des mutations du récepteur au GMCSF, a un traitement spécifique. Le GMCSF est un messager fabriqué par les cellules épithéliales qui active la digestion du surfactant par le macrophage alvéolaire. Dans cette forme, lorsqu’elle progresse malgré les lavages répétés, on peut proposer une greffe de moelle osseuse qui permettra de remplacer les macrophages alvéolaires dont le récepteur est défectueux pas des macrophages sains. Les formes secondaires sont traitées par le traitement de leur cause.
Chez l’adulte, il existe des traitements spécifiques des formes auto-immunes.
Quelles mesures préventives sont recommandées ?
Il n’y a pas de traitement préventif permettant d’éviter la survenue d’une PAP. Dans les formes génétiques, un conseil génétique doit être réalisé pour déterminer si un diagnostic prénatal est indiqué pour une future grossesse. Comme toute maladie respiratoire chronique, les vaccinations de l’enfant doivent être à jour et complétées par la vaccination saisonnière antigrippale annuellement. La protection contre le virus VRS par le Beyfortus® est aussi recommandée les 2 premiers hivers. En cas d’insuffisance respiratoire prolongée associée, rendant l’enfant globalement plus fragile, l’éviction de la garde en collectivité (crèche) est recommandée au moins la première année.